80-90% Faster
Than traditional, manual risk analysis.
AI-Powered Medical Device Risk Analysis. Ready in Hours.
Stop losing weeks to ISO 14971 risk analysis.
SIFo AIRA automates hazard identification, risk estimation, and ISO 14971-compliant documentation for medical devices – delivering structured output in a fraction of the usual time.
Built by MedTech experts and auditors.


















If you’ve worked on a risk analysis before, you know what it actually looks like: redundant discussions for weeks, a spreadsheet growing in every direction, version conflicts, and a document that still gets flagged in the next audit for "inconsistent terminology."
A thorough ISO 14971 risk analysis typically takes a team of 3–6 people between 40 and 150 hours. It’s manual. It’s inconsistent. And it's often the single biggest bottleneck between your device and the market.

AIRA is an AI-powered risk analysis tool developed specifically for medical device manufacturers. It automates the most time-consuming parts of the ISO 14971 process – hazard identification, risk estimation, and control measure documentation – and delivers a structured, first draft in a fraction of the usual time.
What makes AIRA different from generic AI tools: it was built by Simon Föger and Bernhard Lindner, medical device experts who have spent 15+ years reviewing risk documentation from both sides of the audit table. Every output is structured the way auditors expect to see it.
The result: your team starts with a complete, structured baseline – not a blank spreadsheet. A single expert reviews and finalizes. You export a fully structured documentation for your medical device. Done in 2–10 hours.
Traditional vs. SIFo AI‑Assisted
Why industry leaders are switching to automated compliance.
Input Variables
Cost Traditional
0 €
Projected Savings
0 €
per Analysis
Hours Saved
0 h
ROI Factor
0%
*Calculation assumes 5 hours of internal review time.
Transparent costs. High precision. No hidden fees.
For Start-ups and Small Manufacturers
For SMEs including SIFo Expert Support
For Enterprise and High Volume

Simon Föger is a medical device quality expert trusted by manufacturers across Europe – and the mind behind AIRA.
The idea came from a pattern he kept seeing: manufacturers struggling to justify their sample sizes. The problem looked statistical, but the root cause was almost always the same: no solid risk analysis to derive from. Without that foundation, sample size justification becomes guesswork. After helping company after company work backwards to fix their risk management first, Simon realised the gap was universal.
AIRA is his answer: a structured ISO 14971 risk analysis that gives manufacturers the foundation everything else depends on.
As a trainer at TÜV SÜD Academy and consultant to leading MedTech companies, he has reviewed hundreds of risk files and knows exactly what notified bodies and auditors look for.
That regulatory depth is baked into every AIRA output. When you use AIRA, you get Simon's methodology – in a fraction of the time.

Bernhard Lindner is a risk management specialist with over 15 years of hands-on experience in the medical device industry – and the expert behind AIRA's review layer.
As former Head of Quality Management at a medical device manufacturer, Bernhard has lived the full lifecycle of a risk file: from the first hazard brainstorm to the moment an auditor puts it under the microscope. He knows where risk analyses fail, where documentation gaps trigger findings, and what it takes to build a file that holds up – with notified bodies, under MDR, and against FDA 21 CFR 820.
In the SIFo AIRA + Advisory tier, Bernhard personally reviews your AIRA output – you'll get his regulatory judgment to your specific medical device context. He catches the edge cases. He flags the assumptions. He makes the risk file yours. When an auditor asks why you chose that risk acceptance criterion, you will have an answer.
3 Steps to MedTech-Compliant Risk Analysis
"We used AIRA to cross-check the hazards we had already identified, and it immediately became clear how precisely the results fit our product. The tool removes the biggest chunk of work in the initial risk identification phase, and the draft output is a realistic, well-structured starting point for review. For any team starting a new development, it's worth its weight in gold. I'd clearly recommend it."
Benjamin Peschel | Regulatory Affairs | BHS Technologies
SIFo AIRA gives your team AI-powered ISO 14971 documentation in a fraction of the time – developed by a certified auditor and a risk manager who know exactly what regulators expect.
Frequently
Asked
Questions
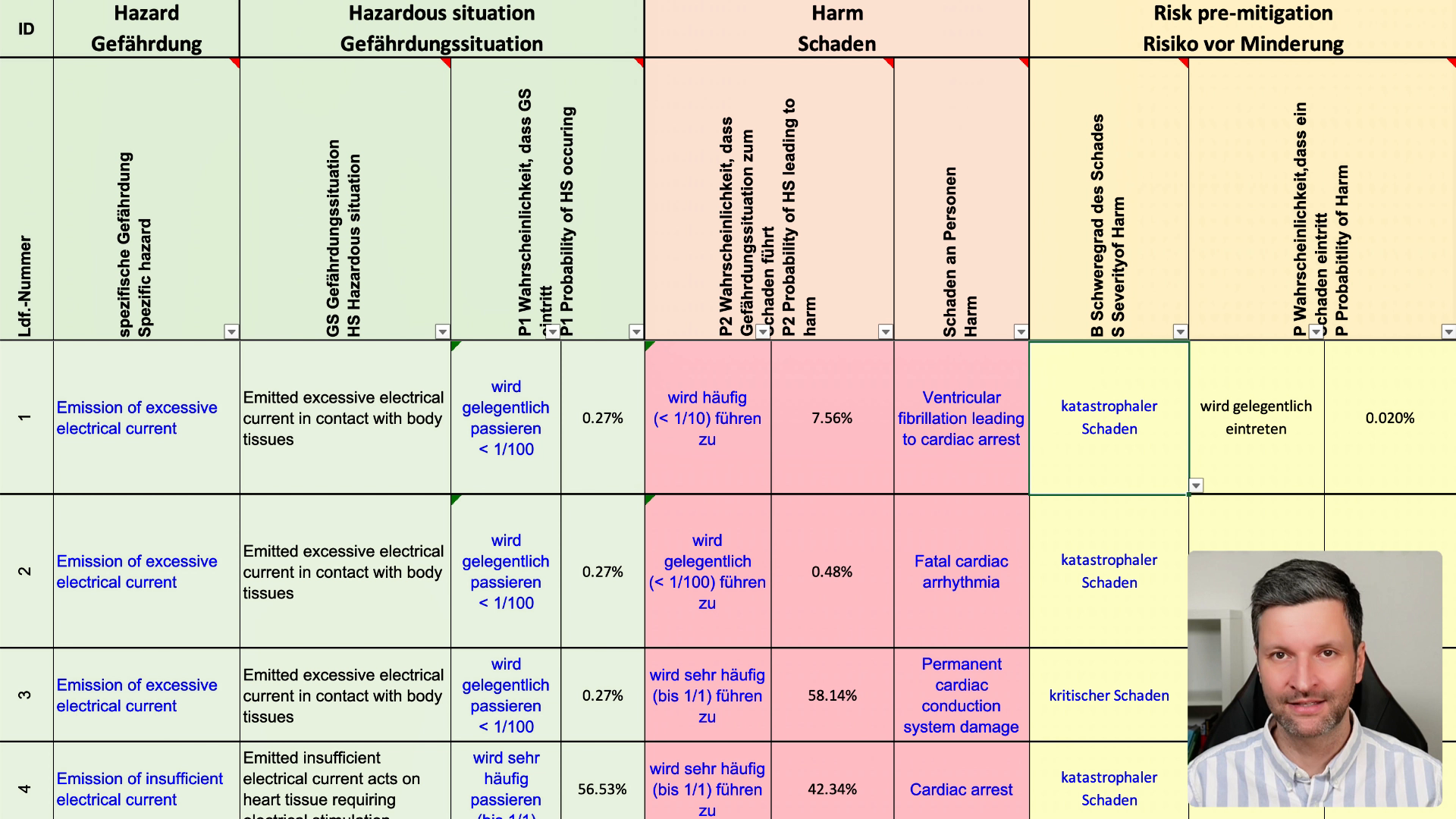
SIFo AIRA delivers a structured, AI-generated risk analysis for your medical device: a complete hazard identification table, hazard situations, occurrence probability estimates (P1 and P2), potential harms by severity, and AI-derived risk acceptance criteria – formatted as a ready-to-use data file. The output is a starting point for your team to review, adjust, and finalize.
AIRA is a service, not a software license. You fill out a structured questionnaire describing your device – intended use, operating conditions, and relevant features. The AI generates your risk analysis and delivers the output to your account. You receive a ready-to-use analysis file, not a platform to manage yourself.
AIRA estimates P1 (probability of a hazardous situation from a given hazard) and P2 (probability that a hazardous situation leads to harm) using AI – drawing on its training data in the same way a human expert draws on experience during a risk moderation session. The AI output is a starting point. Your team then reviews and adjusts probabilities using your own product knowledge, field data, and post-market surveillance results.
The answer is the same as for any manually moderated risk analysis: the team created it. AIRA generates the initial baseline – the same role a pre-filled reference table plays in a manual session. Your team reviews each line, adjusts estimates based on product expertise, and signs off on the final values. The notified body's question is answered by your team's documented review and approval, not by the AI's methodology.
No – AIRA's output is a proposal, not a finished risk analysis. The manufacturer is always responsible for verifying, adjusting, and approving the content before it becomes part of the technical documentation. AIRA replaces the blank spreadsheet and the initial brainstorming session – not the expert review and sign-off process.
Yes. AIRA has been tested with entirely new, fictional device concepts and produced realistic, relevant hazard lists. For genuinely cutting-edge technology where very little published knowledge exists, the output may be more limited – which also reflects the current state of knowledge for any human expert in that situation. For the vast majority of medical device types, AIRA produces a comprehensive and relevant baseline.
AIRA is an AI-powered risk analysis tool developed by SIFo Medical for medical device manufacturers. It automates the ISO 14971 risk analysis process – including hazard identification, risk estimation, and control measure documentation – delivering structured output and reducing documentation time by up to 95%.
Yes. AIRA is not classified as a high-risk AI system under the EU AI Act. It is a decision-support tool – not a medical device itself – and its output requires human expert review before use. A Data Processing Agreement (DPA) is available, documenting which LLMs are used and how the system operates.
Yes. All AIRA output is structured according to ISO 14971:2019 requirements and formatted for compatibility with EU MDR (Regulation 2017/745) and FDA quality system requirements (21 CFR Part 820 / QMSR). Output format is consistent with what notified bodies and FDA inspectors expect to see.
The EUR 3,000 price point can feel steep before you've experienced the traditional alternative. For context: a professional risk management moderator typically charges €150–200/hour, and a complete manual risk analysis with an interdisciplinary team – including physician input for harm classification – often costs EUR 10,000–30,000 in internal and external resources. AIRA delivers the same starting point in a fraction of the time. The ROI becomes most visible once you've been through the manual process at least once.
SIFo AIRA uses AI to generate a comprehensive, structured risk analysis baseline. For the AIRA + Advisory tier, a Risk Management expert from SIFo Medical personally reviews and enhances the output. This human layer is what makes the output audit-resilient, not just compliant.
No. AIRA + Advisory includes expert review from an ISO 14971 risk management specialist, not a clinical physician. For harm severity assessment that requires medical judgment, you will still need input from a clinical expert within your team or network. AIRA's structured output gives them a clear starting point, significantly reducing the time they need to spend on the file.
After completing the device questionnaire (approximately 30–60 minutes), AIRA generates your risk analysis. Total documentation time – including expert review – typically ranges from 2 to 10 hours. This compares to 40–150 hours for traditional manual risk analysis.
Everything in AIRA Essential, plus a 3-hour consultation with Bernhard Lindner, our ISO 14971 risk management specialist. Bernhard reviews your AIRA output in the context of your specific device, flags gaps before your auditor does, and ensures the risk file is structured in a way your QMS can build on. You get the speed of AI and the judgment of an expert who has reviewed hundreds of risk files.
Yes. A full demo video of SIFo AIRA is available on this page – watch it before making any decision. The questionnaire is accessible after creating a free account; output is delivered upon payment.
AIRA is built on a Large Language Model that has developed its knowledge from a broad data base during training. Unlike a search engine, the model does not retrieve individual, citable documents when generating its output – it works much like an experienced consultant who provides assessments based on deep domain expertise. AIRA deliberately does not include automatic source references, because their accuracy and relevance cannot be sufficiently guaranteed in a regulatory context. An incorrect citation would be more damaging in an audit than none at all. This does not affect your ability to defend your risk analysis. AIRA's output is designed as a well-founded starting point – not a finished, submission-ready document. ISO 14971 requires the manufacturer to independently justify every risk decision, regardless of how the initial draft was created. We recommend the following process: Validate the hazards and probability assessments provided by AIRA in a team review. During that review, document the sources your team used for confirmation or correction — for example complaint data, published literature, applicable standards, or clinical data. This documentation is what counts in front of your Notified Body and serves as evidence that the assumptions have been properly verified. The key benefit is that your team no longer starts from zero. The time savings come not from skipping steps, but from having the most labour-intensive step – the first well-founded structure – already in place.
Depending on your tier, you receive: a structured hazard identification table, risk estimation and control measure suggestions, a traceability matrix aligned with ISO 14971, and MDR/FDA-compatible documentation – all in a structured format. For the AIRA + Advisory tier, you also receive an expert-reviewed and enhanced version of the documentation.
Yes – with one important consideration. AIRA delivers a structured ISO 14971 first draft that gives you the foundation your risk management needs to be built on. For a start-up that has never done a formal risk analysis, that structured starting point alone saves weeks of work and prevents the most common mistakes early-stage companies make. That said, someone still needs to review the output and validate it against your specific device context.
If you don't have an in-house QM expert, AIRA + Advisory is the right tier for you. Bernhard Lindner, our ISO 14971 specialist, reviews your output and ensures it is structured in a way your QMS can build on, so you are not left with a document you don't know what to do with. The alternative – hiring an external consultant to build your risk analysis from scratch – typically costs significantly more and takes weeks longer.
Most AI-based risk tools rely on general-purpose AI platforms that are later adapted to the MedTech environment. SIFo AIRA was developed from the ground up by Simon Föger – certified ISO 13485 Lead Auditor, MedTech consultant, and TÜV SÜD Academy Trainer with more than 15 years of hands-on experience in medical device quality management – together with Bernhard Lindner, a recognized expert in medical device risk management. This means the structure, terminology, logic, and traceability of the output reflect real audit expectations and practical regulatory experience from both sides of the table: manufacturer and auditor. With the AIRA + Advisory tier, this expertise is not only embedded in the tool – it is directly applied to your specific risk analysis output.
AIRA is designed for manufacturers of medical devices requiring ISO 14971-compliant risk management, including Class I, IIa, IIb, and III devices under EU MDR, and corresponding FDA device classifications. Contact us if you have a specific device type question before purchasing.
Our Enterprise Subscription plans cover 12–30 analyses per year at significantly reduced per-analysis rates, with priority support. Ideal for manufacturers with multiple products or active development pipelines. Contact us for enterprise pricing.
Contact Us

Book an Appointment
Arrange your initial call with our CEO Simon Foeger
to discuss your project and clarify all your questions.