Medical Device Risk Management: How to Create a MedTech-Compliant Risk Management System According to ISO 14971:2019

Bernhard Lindner

Demystifying ISO 14971: Practical Guidance for Effective Medical Device Risk Management
Many professionals feel overwhelmed when first approaching ISO 14971, as the standard's language can be complex and its requirements seem intricate.
Questions often arise about how to implement the risk management system correctly and how to integrate it with the existing quality management system.
If you share these concerns, you are not alone.
We aim to clarify these interrelationships and provide practical insights to help you establish an effective risk management system that increases patient safety and supports your business objectives.
What is Risk Management According to ISO 14971:2019?
Risk management, as defined by ISO 14971:2019 and mandated by the Medical Device Regulation (MDR), is a well-defined, continuous process designed to identify, assess, and control risks related to medical devices throughout their entire lifecycle.
The primary goal is to minimize risks and prevent harm to patients, users, and third parties.
Personally, I see risk management as a tool that allows us, as medical device manufacturers, to effectively allocate resources. We prioritize activities that are essential to ensuring the safety of our products.
Is Risk Management Required for Medical Devices Under MDR 2017/745?
A common misconception, especially among manufacturers of simple Class I medical devices, is that risk management might not be necessary. Some may think, "What could possibly happen with something like a urine cup?"
However, the MDR is very clear on this matter. It explicitly requires medical device manufacturers to establish, implement, and maintain a risk management system.
This requirement is clearly stated in EU 2017/745 Annex I, Chapter I, Section 3:
"Manufacturers shall establish, implement, document, and maintain a risk management system." [1]
No exceptions are granted.
Therefore, even for seemingly low-risk devices, a lean and efficient risk management system must be in place. Properly designed, such a system integrates seamlessly with your quality management system and supports compliance without unnecessary complexity.
"Manufacturers shall establish, implement, document, and maintain a risk management system."
What Are the Regulatory and Normative Requirements (as of 2025)?
In recent years, there have been significant changes in the regulatory and normative requirements for risk management in medical devices.
For the European market, the MDR (EU) 2017/745 (Annex I, Chapter 1) mandates that manufacturers must operate a risk management system.
The ISO 14971:2019 standard (harmonized with MDR (EU) 2017/745 in EN ISO 14971:2019/A11:2021) outlines what needs to be done to comply with regulatory risk management requirements for medical devices.
The ISO/TR 24971:2020 provides guidance on how to implement these requirements in accordance with the MDR and ISO 14971:2019.
Key Benefits of Medical Device Risk Management
Operating a risk management system requires both financial and organizational effort. That’s why we are often asked: "What value does risk management actually bring to the company?"
In some companies, you’ll hear responses like: "It doesn’t add any value. Nothing ever goes wrong with our product anyway. Honestly, we just create the risk management documents at the end of the project just to have something to show the auditor."
This is essentially self-deception. I find this particularly unfortunate because this approach only wastes paper and expensive working hours without providing any real benefit.
With a comparable level of effort, we could do it properly – improving product safety while also reducing costs.
Benefit 1: Enhanced Safety
The foremost benefit of risk management is protecting patients, users, and third parties from harm caused by medical devices. By systematically identifying hazards and controlling associated risks, manufacturers can significantly reduce the likelihood of adverse events. But that's not the only benefit.
Benefit 2: Efficient Resource Allocation
Risk management helps us allocate limited resources effectively, focusing on the most critical measures. It also forces us to identify and implement these measures early in the project. This shifts error correction to the phase where product defects originate, reducing or even preventing costly late-stage fixes such as recalls. In this way, risk management pays for itself. The second objective of risk management is to prioritize resource usage, ultimately saving costs.

Benefit 3: Protection Against Product Liability
We must not forget that damages related to a medical device can lead to product liability claims. In such cases, the documentation of risk management activities serves as crucial evidence. If risk management is not conducted properly, is delayed, or lacks traceability, it could be interpreted as gross negligence. Effective risk management protects both the company and the individuals responsible for product safety from serious consequences in the event of liability claims.
Benefit 4: Driving Innovation
When incidents occur more frequently than acceptable, solutions are needed to reduce risk. This necessity can lead to new technical advancements or even entirely innovative products. Risk management drives innovation, creating competitive advantages in the market.
Common Mistakes in Medical Device Risk Management
Regulatory and normative requirements for risk management are highly complex, and mistakes can have serious consequences. It’s no surprise that those responsible for risk management often feel uncertain and fear making errors in the system.
Additionally, risk management systems tend to evolve over time rather than being systematically designed from the start. As a result, they often become overly complex, redundant, and difficult to navigate.
Many top-level executives fail to recognize how powerful an effective risk management system can be. Consequently, those responsible often lack the necessary resources to build and maintain a system that provides real value beyond just fulfilling documentation requirements.
This not only wastes resources but can also lead to nonconformities in audits and inspections, or even costly legal consequences in the event of an incident.
Here are 10 common mistakes that make risk management systems unnecessarily complex, expensive, and ineffective:
1. Lack of Integration with Quality Management System: Risk management should not exist in isolation but should be seamlessly linked to the quality management system (ISO 13485:2016) to ensure coherence and effectiveness.
2. Missing "Sense of Urgency": Without top management’s commitment, risk management often lacks the necessary priority.
3. Inappropriate Tools: Using unsuitable risk management tools or software can lead to inefficiencies and unreliable results.
4. Unclear Distinction between Risk Analysis and FMEA: Without clear boundaries, these processes overlap, causing inefficiencies in your risk management.
5. Unclear Responsibilities: Without defined ownership, risk management tasks may be neglected or inconsistently performed. Effective risk identification processes and clear assignment of responsibility are crucial to ensure all identified risks are managed.
6. Insufficient Training: Employees responsible for risk management activities lack proper education and training.
7. Unjustified Acceptance Criteria: Risk analysis and FMEA acceptance thresholds are not logically derived or supported by data.
8. Redundant Activities: Duplicate tasks waste time and resources.
9. Incomplete Specifications: Missing or vague specifications increase uncertainty and risk.
10. Ambiguous Requirements: Guidelines and instructions are not clearly formulated, leading to misinterpretation and errors.
Avoiding these pitfalls helps create a lean, compliant, and effective risk management system that supports product safety and regulatory compliance.
How to Build a Successful Risk Management System According to ISO 14971:2019
Now that we have covered the most common mistakes, let's explore the key factors that contribute to successful risk management.
Risk management doesn’t happen on its own. This is why regulations require us to establish, implement, and continuously improve a structured system for it. At its core, risk management involves analyzing, assessing, and controlling risks.
A risk management system directly or indirectly affects nearly all areas of a medical device manufacturer and every stage of a product’s lifecycle. That’s why it’s crucial to build a well-structured and thought-out system to mitigate risks.
But how can you organize a risk management system so that it remains practical in daily operations?
To establish a successful risk management system, medical device companies should focus on the following 8 key success factors:
1. Appoint a Qualified Person: Designate someone responsible for developing, operating, and maintaining the risk management system.
2. Establish Management Commitment: Ensure top leadership understands and supports the importance of risk management.
3. Define a Clear Risk Policy: Communicate the company’s approach to risk management to all employees.
4. Specify Required Activities: Document what needs to be done in the risk management process through procedural instructions.
5. Define How Activities Should Be Performed: Provide work instructions, templates, and forms to guide employees.
6. Use Only Necessary Tools: Avoid unnecessary complexity by selecting appropriate risk management tools.
7. Train and Qualify Staff: Ensure employees involved in risk management are properly trained.
8. Monitor the Risk Management System: Use key performance indicators (KPIs) to track effectiveness and drive continuous improvement.
By implementing these factors, companies can build practical, efficient, and compliant risk management systems that integrate well with their quality management systems.
Attention, CEOs: This Is the Biggest Misconception in Medical Device Risk Management
I often work with companies that have repeatedly failed to implement risk management, risk analysis, or FMEA. With each unsuccessful attempt, frustration grows until these tools become completely "burned out".
At that point, simply mentioning them triggers eye-rolling and resistance among employees. Understandably so – by then, risk analysis and FMEA are associated with negative experiences.
So how can we successfully integrate risk analysis and FMEA as an essential part of the system?
One key factor – in my opinion, the decisive factor – is the understanding of top management.
I frequently see that executives have little to no knowledge of risk management systems for medical devices. To them, risk management seems like a necessary evil – costly and without real benefit.
To successfully implement risk analysis and FMEA, the first and most crucial step is ensuring that top management understands the value and benefits of risk management.
The Underestimated Risk Policy: Theory and Practice
The risk policy is one of the most underrated documents in risk management. Its primary purpose is to provide employees with a framework to guide their decisions.
However, critical mistakes often occur in its implementation.
One common issue is that top management is not fully convinced of the importance of risk management. As a result, the risk policy is seen as just another document – something to pin on the wall to satisfy auditors, rather than a meaningful guideline.
But if top management doesn’t truly believe in the necessity of risk management or how it should be implemented, then risk management will remain nothing more than an expensive exercise in paperwork.
To avoid this, ensure that top management is genuinely committed to risk management and the risk policy.
For a risk policy to be effective, it must be consistently communicated and actively practiced. Employees will only take it seriously if they see leadership demonstrating it in action.
At the very least, integrate the risk policy into annual awareness training and align key decisions with its principles.
Even better: embed the risk policy into daily operations and regularly remind employees of its importance.

The complex tasks of risk management cannot be handled with just one tool. Using only FMEA or only risk analysis is not enough. Medical device risk management requires a toolbox of methods tailored to different tasks.
Annex B of ISO/TR 24971:2020 lists several risk management tools – FTA, ETA, HAZOP, PHA, and FMEA, among others – that should be selected based on suitability for the task at hand.
However, selecting the right tools can be tricky. Using an inappropriate tool for analysis can lead to significant extra effort and unreliable results.
Which risk management tools are the right ones?
It depends on the task. Not every tool listed in ISO/TR 24971 is suitable for every aspect of risk management. Think of it like a toolbox – you wouldn’t use pliers to drive a screw or a saw to hammer in a nail.
Similarly, FMEA is not the right tool for identifying risks, while a risk analysis is not ideal for determining root causes.
Choosing the right method for the right task is key to an efficient and effective risk management system.
The following section presents essential risk management tools, outlining their purpose and how they are applied in the context of medical device risk management.
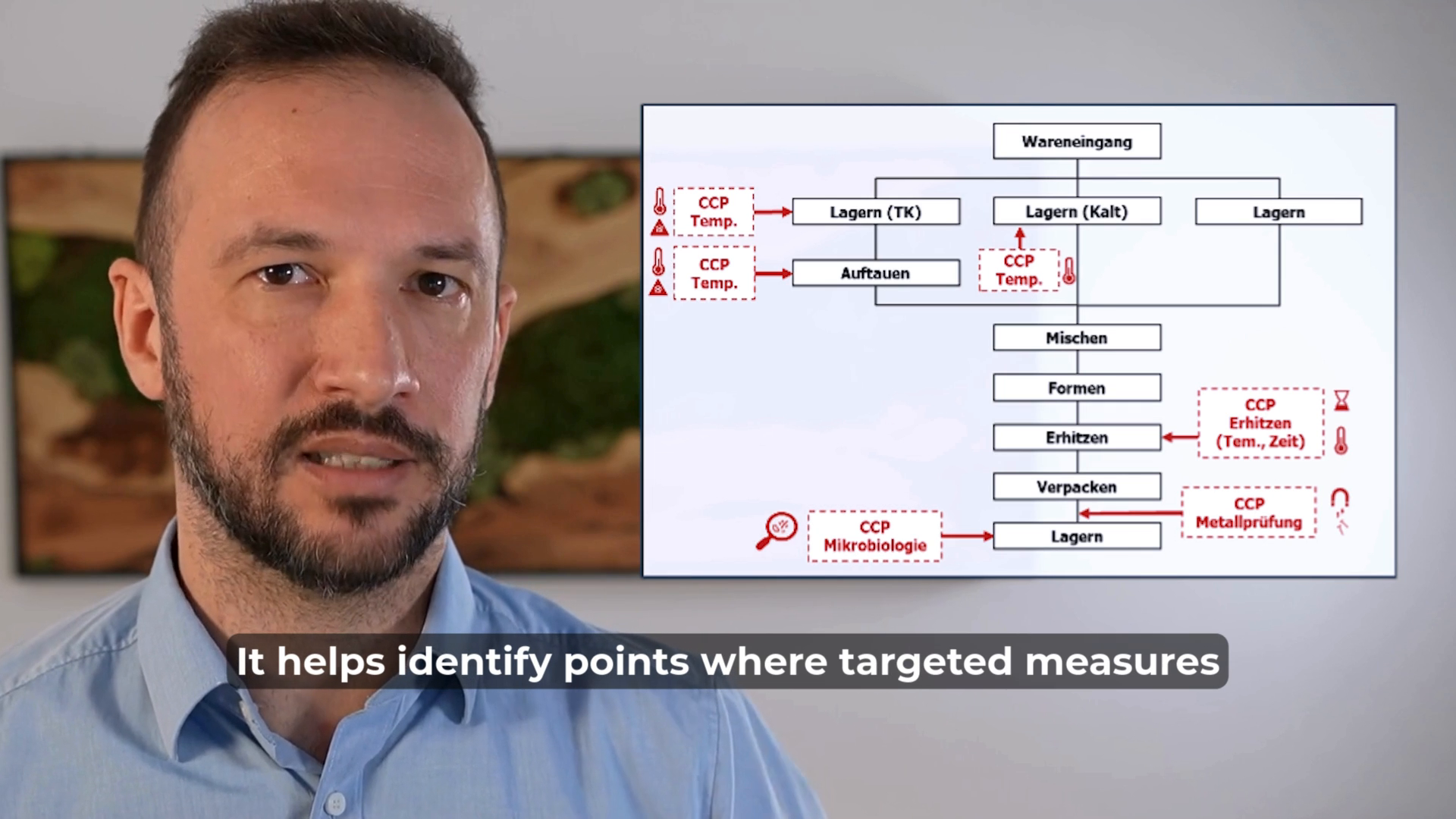
HACCP (Hazard Analysis and Critical Control Points) is a useful method for identifying hazards and root causes in the manufacturing process.
By pinpointing critical control points (CCPs), we can implement measures to keep these hazards under control.
HACCP is widely used in food safety risk management, but it is rarely applied in the medical device industry – despite its potential benefits. Implementing HACCP in medical device manufacturing could help improve process control and risk mitigation in production.


HAZOP (Hazard and Operability) is a powerful tool for systematically identifying hazards caused by functional failures in a system or subsystem. It uses guide words (such as no, none, too much, too little, too late, etc.) to derive potential failure states of functions.
HAZOP is often used during the development process as a complementary tool to hazard analysis, helping to uncover risks that might otherwise be overlooked.
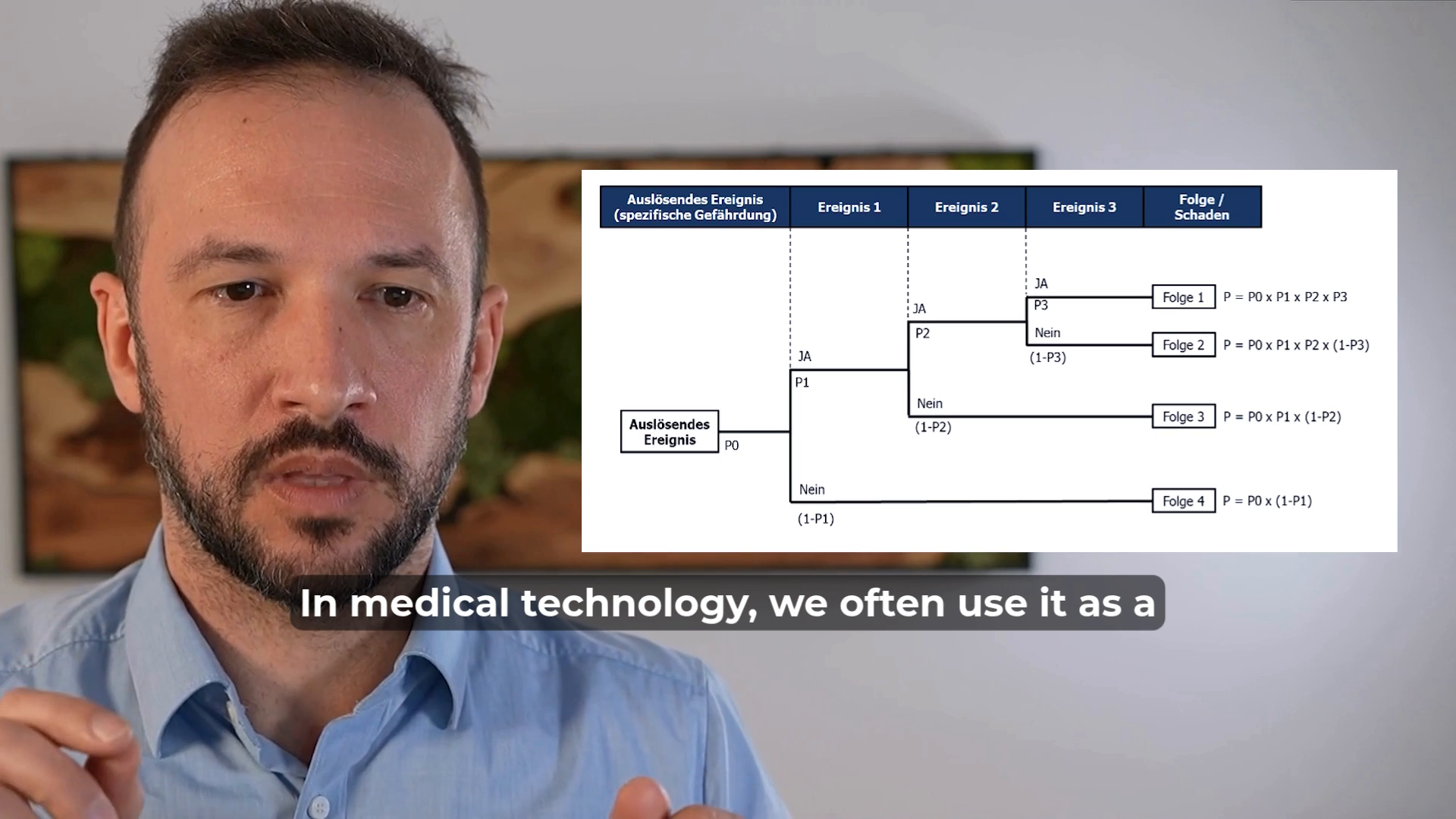
FTA (Fault Tree Analysis) is a valuable tool for identifying the causes of a known effect and analyzing their dependencies.
It uses Boolean logic (AND/OR gates) to map out how different causes interact and contribute to an event.
In the medical device industry, FTA is frequently used in root cause analysis, helping to systematically trace failures back to their sources and understand their interconnections.

ETA (Event Tree Analysis) is a method used to evaluate the sequence of events following an initiating incident and to assess the potential outcomes.
In the medical device industry, ETA is often used as a complementary tool in risk analysis. It helps identify "reasonably foreseeable" chains of events that could lead to harm and allows for the calculation of their probabilities.

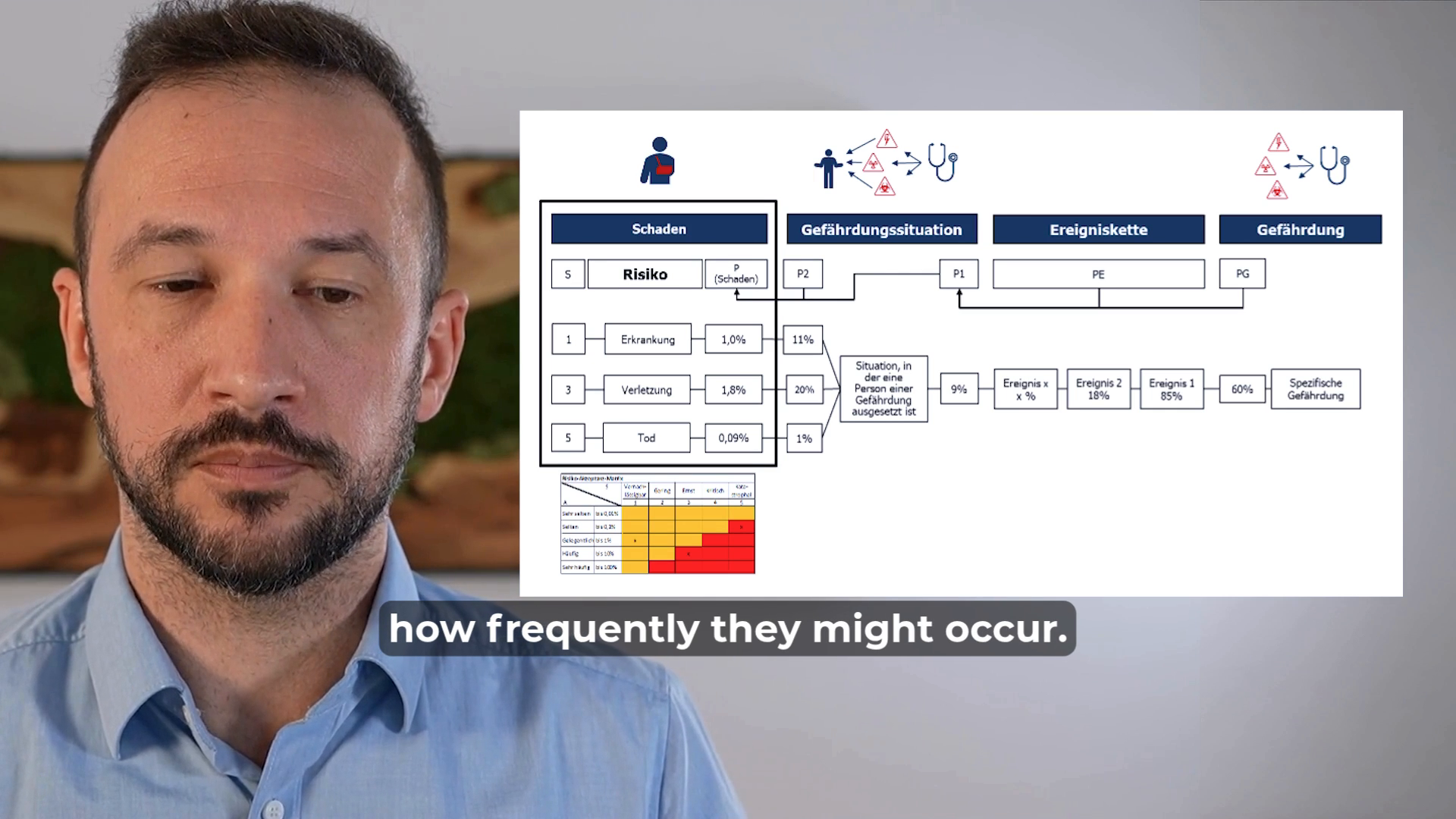
Risk analysis is a tool used to evaluate the impact of product defects.
We assess what harm to individuals could result from the functions and characteristics of a medical device and estimate both the severity and likelihood of these harms occurring.
To determine whether the frequency of a particular harm is socially acceptable, we compare it against a predefined acceptance threshold.
However, risk analysis is not designed to identify the root causes of product defects – its focus is on the effects rather than the causes.
In the medical device industry, risk analysis is a core tool in risk management.
⚠ Beware: Risk analysis is often mistaken for FMEA, but they serve different purposes.


Creating a risk analysis from scratch typically ties up 3–6 people for 40–150 hours.
SIFo AIRA – AI Risk Analysis delivers your structured ISO 14971 first draft in 2 hours – complete with hazard identification, P1/P2 probabilities, and risk acceptance criteria. Your team reviews it, you own it.
FMEA: Failure Mode and Effects Analysis
FMEA (Failure Mode and Effects Analysis) is a tool used to analyze where a product defect originates.
By evaluating a combination of severity of the effects, detectability, and likelihood of occurrence, we determine the priority of corrective actions.
⚠ Beware: FMEA alone does not fulfill the regulatory and normative requirements for risk analysis!
The optimal risk management approach is achieved by combining FMEA with risk analysis.

The 7 Steps of Risk Management
Risk management doesn’t have to be complicated or overwhelming. Risks can also be managed effectively with simple methods. Stick to the fundamental principles, and you’ll be on the right track.
Effective medical device risk management follows a structured seven-step process:
Step 1: Determine Risk Acceptance Criteria
Before you begin analyzing risks, you need to establish thresholds for acceptable risk related to your product. To put it bluntly: "How many injuries or fatalities are considered acceptable?"
It is crucial to define these acceptance criteria at the very beginningto create a reference point against which risks can be measured.
Risk acceptance criteria depend on regulatory requirements and the benefit of the product. They must be determined, justified, and documented.
Step 2: Plan Risk Management Activities
Carefully plan the risk management activities you intend to carry out. Ensure that only activities relevant to the project, product, and manufacturing process are included.Consider interfaces with other companies and determine who is responsible for maintaining the risk management file.
⚠ Important: Companies with manufacturer responsibility may need access to risk management documents, such as FMEAs from suppliers.
Document the acceptance limits for residual risks and the overall residual risk.Plan when risk management activities need to be carried out within the project.
Keep in mind: Risk management helps identify critical errors early, allowing you to address them at the beginning of the project – ultimately saving costs.
Ensure that planned risk management activities are carried out on time.
Make sure that these activities are integrated throughout the project rather than being delayed until the end of development or a change process. This approach ensures that the necessary efforts in risk management provide the greatest benefit for both product safety and the company.
Additionally, document activities at the time they are performed. Proper documentation serves as evidence of compliance and can be crucial in case of product liability claims.
Step 3: Analyze Risks
First, identify the hazards associated with your product, determine under what circumstances individuals may be exposed to these hazards, and assess the potential harm that could result.
Then, estimate the probability of occurrence and the severity of the harm.
Finally, calculate the maximum acceptable occurrence rate of products with the identified hazard, considering the predefined risk acceptance criteria for harm occurrence (risk acceptance limit).
Step 4: Identify Risk Control Measures
For each hazard identified in the risk analysis, examine its causes in terms of usage, design, and manufacturing.
Define preventive and detection measures in both design and production to minimize the occurrence of these hazards as much as possible.
Ensure that evidence of implementation is properly documented to demonstrate compliance and effectiveness.
Step 5: Verify the Effectiveness of Measures
Verify that the implemented measures reduce the occurrence of hazards to an acceptable level. Leverage existing quality management processes, such as usability studies, design verification, and process validation.
Ensure that the determined maximum acceptable occurrence rate for each hazard is considered when calculating sample sizes for verification testing.
⚠ If verification or validation fails to demonstrate that the occurrence rate is below the acceptable threshold, it is likely that the risk acceptance limit is exceeded, meaning that the hazard may occur more frequently than is considered acceptable.
Step 6: Evaluate Residual and Overall Risks
Assess each residual risk and verify that it complies with the acceptance limits documented in the risk management plan.
If it is not possible to reduce residual risks below these thresholds, the benefit-risk ratio must be evaluated. Also, make sure to check whether new risks have emerged as a result of the implemented measures.
Ensure that all documented residual risks are included in the Instructions for Use (IFU).
This is especially important for demonstrating, in the event of product liability claims, that patients were properly informed about potential risks and side effects.
Evaluate the overall residual risk against the acceptance limits documented in the risk management plan.
Step 7: Risk Management Report
Summarize all risk management activities in a report and discuss the results.
Ensure that all planned activities have been completed, and the effectiveness of risk control measures has been verified.
Make sure the report is clear, concise, and meaningful.
Tip: In audits and inspections, risk management reports are often the starting point for evaluating the entire risk management process. A well-structured report can significantly improve transparency and compliance.

About the Author
Bernhard Lindner, MSc. is a recognized expert in quality and risk management for medical devices, with over 15 years of experience. As the former Head of Quality Management at a medical device manufacturer and now an independent consultant, he supports companies in the efficient and compliant implementation of legal requirements in accordance with ISO 13485, ISO 14971, MDR, and FDA 21 CFR 820. His expertise lies in risk management, especially risk analysis and FMEA facilitation – practical, results-oriented, and focused on compliance and efficiency. Having worked with leading companies in the medical and automotive sectors, he combines in-depth technical knowledge with practical experience. For those seeking professional risk management and quality optimization, Bernhard is a trusted partner for long-term, sustainable success.

Risk-Based Samples Sizes
Learn how to justify sample sizes using a clear, risk-based and statistically sound approach that reduces validation effort and cost.
Gain a practical framework you can confidently defend in audits across TMV, design verification, packaging, and process validation.
Further helpful links and resources:
Newsletter: Join our community and be the first to receive updates and news.
Free Resources: Get free access to checklists & templates.
TMV Guide: Your practical guide to perform test method validation (incl. templates & videos).
TMV Online Course: Become an expert in Test Method Validation.
SIFo AIRA – AI Risk Analysis: Use SIFo AIRA to create your risk analysis within hours, instead of weeks – structured, fast, and MedTech-compliant.
References
[1] European Union, Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, Annex I, Chapter I, Section 3, Official Journal of the European Union, L 117, pp. 1–175, May 5, 2017. [Online]. Available: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32017R0745
[2] International Organization for Standardization, ISO 14971:2019 – Medical devices – Application of risk management to medical devices, Geneva, Switzerland: ISO, 2019.
[3] International Organization for Standardization, ISO/TR 24971:2020 – Medical devices – Guidance on the application of ISO 14971, Geneva, Switzerland: ISO, 2020.